









Various congenital diseases affect the heart, with some involving multiple organ systems and encompassing multiple presentations. The broad literature on the subject of such syndromes describes various novel syndromes with presentations not seen previously. Our case is one such presentation, where the constellation of findings has not been reported before, nor do they satisfy the diagnostic criteria of any existing syndromes.
Case Report
A 24-year-old man, born of healthy non-consanguineous parents, presented with shortness of breath on walking half a mile and a 9-month history of palpitations, along with a cough with bouts of haemoptysis for 6 months. Developmental milestones were normal. His father died of a stroke at the age of 60, but his mother and six siblings (three men and three women) were well. He was treated with furosemide by his local doctor.
On examination, his weight was 40 kg and height 150 cm. He had a receding chin, a rudimentary right ear with normal auditory function (Figure 1), bilateral cataract, a drooping right upper eyelid and mild central cyanosis. Other significant findings included digital clubbing and polydactyly of the right thumb (Figure 2).
Vital signs were normal with a pulse of 84 BPM and blood pressure of 130/80 mmHg. Cardiovascular examination revealed raised jugular venous pressure, apex beat localisation to left fifth intercostal space, no pitting oedema, normal first and second heart sounds and a soft systolic murmur in the second left intercostal space. Respiratory auscultation was normal. Mini Mental State Exam and neurological examination were normal. Fundoscopy was difficult to perform because of bilateral cataracts, but did not reveal any obvious abnormality. Abdominal and genital examinations were normal.
Chest X-ray revealed enlarged pulmonary arteries with peripheral pruning and an elevated cardiac apex due to right ventricular hypertrophy. ECG suggested features of right heart hypertrophy and frontal plane QRS right axis deviation. Ultrasonography of the abdomen was normal. Echocardiography revealed biatrial enlargement, biventricular hypertrophy, a large ostium secundum atrial septal defect and a peri-membranous ventricular septal defect, each measuring approximately 18 mm with bidirectional flow shunting predominantly from right to left. Severe pulmonary arterial hypertension was present with moderate tricuspid regurgitation. Pulmonary venous drainage was normal. Blood tests showed mild erythrocytosis with elevated haemoglobin. Urea and electrolytes, liver and thyroid function tests were normal. Karyotyping was performed, which was normal (46 XY).
The patient was offered cardiac catheterisation, which he declined. Conservative management was followed and his diuretic dosage was increased. After discharge, he was followed up over the next year. He remained adherent to medications and his symptoms improved.
Discussion
The patient had Eisenmenger syndrome with multiple upper limb and facial abnormalities along with cataracts, suggestive of a hereditary disorder. The diagnostic dilemma stemmed from the difficulty in unifying the clinical findings under one particular syndrome. Our findings bore resemblance to – but did not satisfy the diagnostic criteria for – existing syndromes. Hence, we narrowed down our differential diagnoses to the clinical entities described in the following paragraphs.
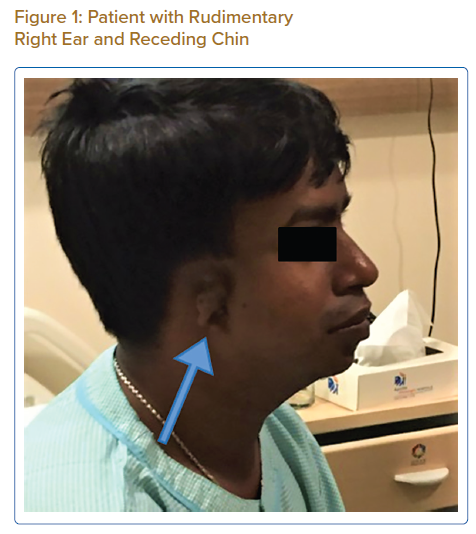
Congenital heart disease, ear deformity and polydactyly in our patient in line with the diagnosis of Patau syndrome, which involves trisomy of all or part of chromosome 13. However, our patient lacked other characteristic findings of Patau syndrome, such as mental retardation, umbilical hernia, cleft lip, coloboma and a congenital renal anomaly.1 In addition, survival until the third decade of life is extremely uncommon in Patau syndrome and when it occurs it is usually associated with mosaicism.2
Skeletal abnormalities of the upper extremities, along with cardiovascular anomalies raised the suspicion of Holt-Oram syndrome. However, the findings, such as facial deformities and cataracts and the absence of cardiac conduction defects or a family history of congenital heart disease, did not support this diagnosis.3
Smith-Lemli-Opitz syndrome is suspected in patients with distinctive facial features, microcephaly and intellectual disability along with cardiac, lung, renal or digital abnormalities.4 Apart from growth restriction and feeding difficulties, with features of autism, Smith-Lemli-Opitz syndrome has been associated with a characteristic atrioventricular canal defect.5
The term CHARGE syndrome encompasses coloboma, heart defects, choanal atresia, retarded growth and development, genital abnormalities and ear anomalies. However, our patient did not satisfy any of the three major criteria (Verloes updated criteria) for this diagnosis.6
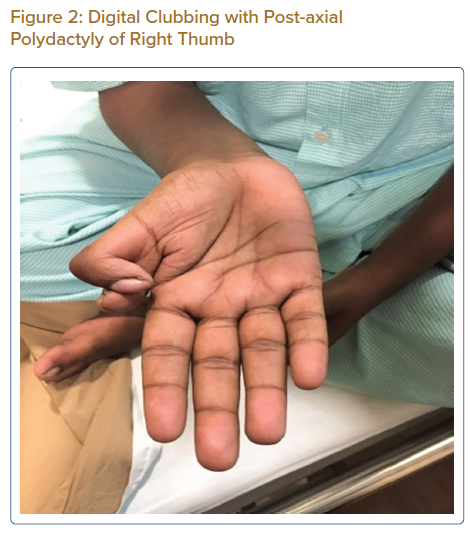
Levin et al. described four patients with atrioventricular septal defects and postaxial polydactyly, without the presence of any other major deformity.7 These cases resembled Ellis-van Creveld syndrome, which includes short limb dwarfism, polydactyly, abnormal development of fingernails, normal IQ and motor development and congenital heart defects in over half of the cases.8 Our patient bears a strong resemblance to these cases, with the coexistence of cardiac abnormalities and upper limb deformities, albeit the facial deformity and early cataracts remain unexplained.
Hence, our patient may present a variant of Ellis-van Creveld syndrome, although the exact diagnosis is uncertain.
Apart from karyotyping, no detailed chromosomal analysis could be carried out on this patient. This lack of detailed chromosomal analysis makes it impossible to make a definite diagnosis, which is a limitation of this case report. The question of whether our case is the first reported case of its kind, or a phenotypic variant of an already reported syndrome, remains unanswered.