









Heart failure (HF) is a clinical syndrome that represents the final manifestation of many underlying cardiovascular disorders and is the most common cause of hospitalisation in the elderly population.1 The number of patients with HF has been increasing rapidly over the past few decades in Japan, mainly because of the ageing population.2,3 Epidemiological transition and population ageing are no exception in Asian countries, and HF is expected to become a larger epidemic in the near future.4
Despite decades of therapeutic development, cardiovascular disease remains the leading cause of morbidity and mortality globally, accounting for an estimated 18.6 million deaths annually in the US.5 In Japan, the 3-year mortality associated with cardiovascular death in hospitalised patients with HF is approximately 20%.6 In a study that compared the changes in the outcomes of Japanese patients with HF between the 2000s and the 2010s, the rate of rehospitalisation for patients with both HF with preserved ejection fraction (HFpEF) and HF with reduced ejection fraction (HFrEF) significantly decreased in the 2010s; however, the cardiac mortality rates were not significantly reduced in either HFpEF and HFrEF.7
In recent decades, numerous clinical trials for HF have revolutionised the management and understanding of its pathophysiology.8 In the 1980s, angiotensin-converting enzyme inhibitors (ACEIs) were proven to be effective for the treatment of various functional classes of HF, demonstrating the importance of neurohormone factors in HF. In the late 1990s, mineralocorticoid receptor antagonists (MRAs) and β-blockers were identified as two drug classes with a great impact on mortality in patients with HFrEF. The combination of ACEI, MRA, and β-blocker reigned as a cornerstone therapy for patients with HFrEF until the 2010s.
In the 2000s, implantable devices such as ICDs and CRT produced a reduction in mortality as well as an improvement in the quality of life in patients with HFrEF.9 And over the last two decades, mechanical circulatory support and left ventricular assist devices (LVADs) have altered the landscape of HF management and are now used as either a bridge to transplant (BTT) or as an alternative to heart transplantation (destination therapy: DT).10 Superior outcomes with the newest, third-generation LVAD (a fully magnetically levitated centrifugal-flow LVAD) have been observed, particularly with respect to blood compatibility-related adverse events, including pump thrombosis and stroke.11 In addition, the superior treatment effect of third-generation LVAD was similar in patients with BTT or DT.12
In the 2010s, new classes of drugs, such as angiotensin receptor–neprilysin inhibitor (ARNI), sodium-glucose cotransporter 2 inhibitors (SGLT2Is), and ivabradine have gained attention.13–15 The combination of ARNI, MRA, β-blocker and SGLT2I is now recognised as the ‘fantastic four’ or ‘foundation therapy’, which has been shown to improve the prognosis of HF compared with conventional therapy and is now incorporated into the guidelines of HF therapy.16–20 Recently, two other classes of drug have been tested in clinical trials. Vericiguat, a soluble guanylate cyclase stimulator, has been shown to reduce cardiovascular death and hospitalisation for HF in the Victoria trial, while omecamtiv mecarbil, a selective cardiac myosin activator, reduced the primary outcome in the GALACTIC-HF trial.21,22 Furthermore, drugs for specific heart diseases, such as mavacamten for hypertrophic obstructive cardiomyopathy or tafamidis for transthyretin amyloid cardiomyopathy, are also expected to reduce HF associated with these diseases.23–25
Despite the long history of the development of therapeutic approaches for HF described above, significant knowledge gaps remain regarding robust and specific biomarkers for risk prediction, comprehensive understanding of the pathogenesis of HF, and critical regulators of HF development. A more comprehensive and systematic understanding of HF through basic research is indispensable for significant breakthroughs in its treatment.
Research on Heart Failure
Mechanism of Heart Failure
HF has many potential causes, including ischaemia, haemodynamic overload, ageing, inflammation and differentiation abnormalities. In many cases, an intricate combination of multiple risk factors triggers HF.26 Although the pathogenesis of HF is complex, we have elucidated some aspects of the underlying molecular mechanisms through basic research.
Heart Failure and Cardiac Hypertrophy
Cardiac hypertrophy is the primary sign of HF. Pathological cardiac hypertrophy causes HF owing to inadequate signalling, leading to unfavourable remodelling. The most common causes of cardiac hypertrophy are hypertension, MI, valvular disease, and genetic mutation (hypertrophic cardiomyopathy; HCM). As the disease progresses, cardiac contractility decreases, occasionally resulting in HF; however, the mechanism of collapse is still unclear.
Several pathways induce pathological hypertrophy. It has long been known that transforming growth factor-β/Smad signalling is involved in pathological hypertrophy.27 In addition, various factors, such as angiotensin II, endothelin-1, mechanical forces and catecholamines, can activate intracellular hypertrophic signalling pathways. The calcineurin–NFAT (nuclear factor of activated T-cells) pathway also causes hypertrophy by increasing fetal gene expression.28 Thus, the activation of various signalling pathways has been shown to be involved in the pathological response process, which provides evidence for the use of several clinical therapeutic agents that manipulate neurohormone factors regulating the signalling pathways responsible for HF.
We focused on elucidating the molecular mechanism underlying this transition from compensated to decompensated hypertrophy. One of the conventional features of decompensated hypertrophy is the occurrence of adverse changes in tissue oxygenation, such as the suppression of angiogenesis, endothelial dysfunction, and fibrosis of myocardial tissue. In hypertrophic hearts, increases in sarcomere synthesis and cardiomyocyte volume result in hypoxia of myocardial tissue due to the relative lack of vasculature and oxygen supply. Although hypoxia is usually ameliorated by induction of angiogenesis via activation of hypoxia-inducible factor (HIF), angiogenesis is suppressed in failing hearts by elevated expression of p53, which inhibits and degrades HIF. As a result, myocardial tissue is unable to evade ischaemia and contractility is reduced. These abnormalities are restored by inhibition of p53 (Figure 1).29
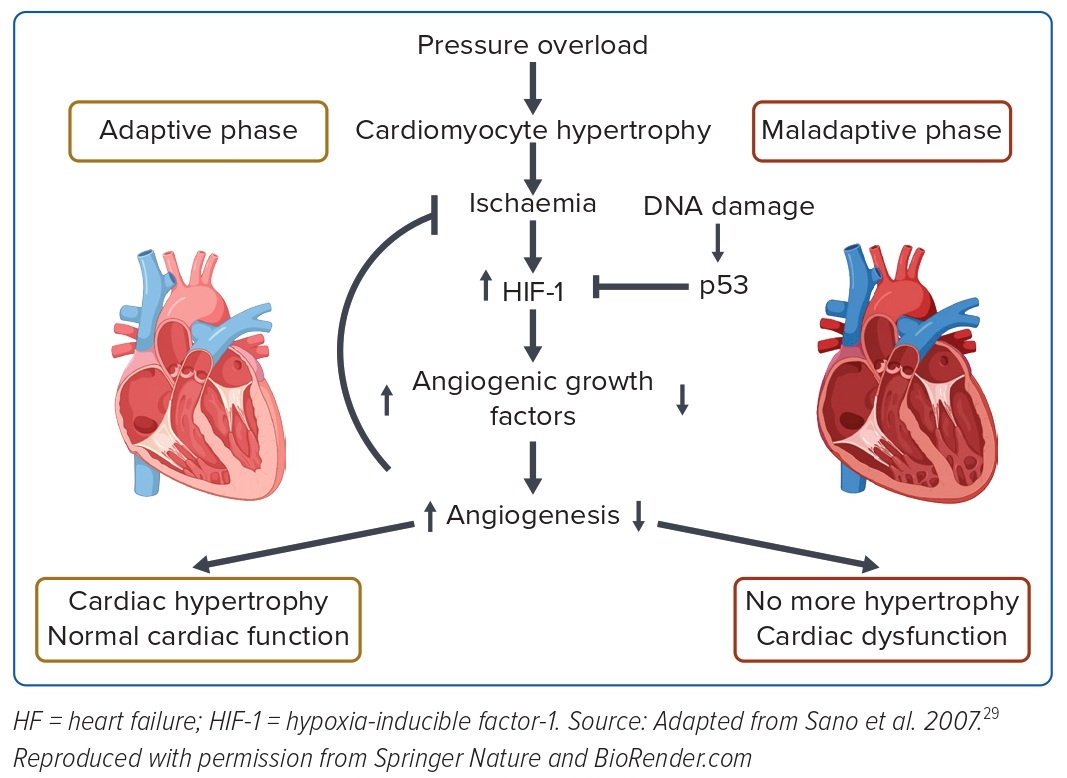
Subsequently, a number of studies have focused on the transition to pathological systolic dysfunction from hypertrophy and have reported the involvement of a variety of factors, including immune cells, hydrogen sulfide signalling, extracellular matrix remodelling, as well as hypoxia-related pathways.30–32 Many attempts are underway to identify signals, enzymes and molecules other than those targeted by existing therapeutics to prevent hypertrophy-related decompensatory failure.
Heart Failure and DNA Damage
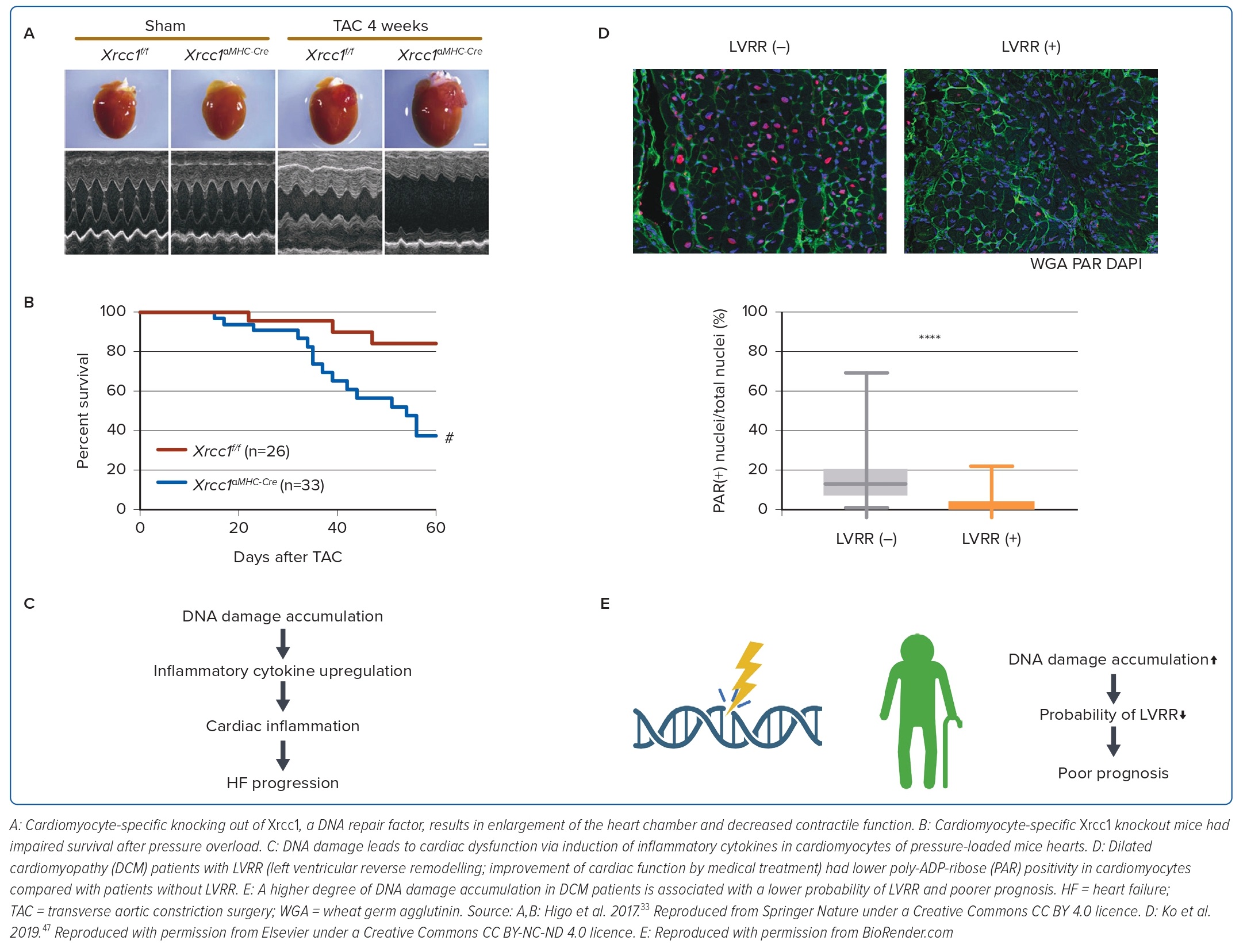
After determining the unfavourable effects of p53 activation in failing hearts, we focused on the association between the accumulation of DNA damage, a major cause of p53 activation, and cardiac dysfunction. In a subsequent study, we generated and analysed a mouse model of pressure overload HF, in which comet assays showed that DNA single-strand breaks accumulated in cardiomyocytes.33 Cardiomyocyte-specific knockout mice of Xrcc1, a DNA repair protein (αMHC-Cre tg/-; Xrcc1 flox/flox) had a higher degree of DNA damage accumulation in cardiomyocytes after pressure overload and a higher mortality rate (Figure 2A,B). Furthermore, when these mice were crossed with Atm knockout mice, an initiator of the DNA damage response (DDR), the expression of inflammatory cytokines was reduced and cardiac dysfunction was suppressed. These results indicate that the accumulation of DNA damage in the heart is the cause of HF and suggest the possibility of suppressing cardiac dysfunction through DDR inhibition (Figure 2C).
The accumulation of DNA damage is a major cause of cellular senescence. Furthermore, senescent cells acquire the ability to secrete specific cytokines, chemokines and matrix-remodelling proteases, known as senescence-associated secretory phenotypes, which negatively affect tissue function. Multiple reports indicate that the long-term accumulation of senescent cells impairs cardiac function and promotes heart disease.34 In contrast, removal of senescent cells has been shown to improve cardiac function in a continuous angiotensin II infusion model, suggesting that senescence due to DNA damage is a cause of HF.34
Recently, various groups have reported that accumulation of DNA damage in cardiomyocytes underlies the pathogenesis of HF. Zhang et al. reported that calmodulin-dependent kinase (CaMK) IIδ, a splice variant of CaMKII, is abundantly expressed in the heart and that this isotype protein causes DNA damage accumulation by inhibiting DNA damage repair through degradation of the ubiquitin-conjugating enzyme (UBE2T).35 Sato et al. showed that Caren, a cardiomyocyte-specific lncRNA, maintains cardiomyocyte function by suppressing DDR and activating mitochondrial respiration through the repression of Hint1 expression.36
Zhao et al. reported that the level of circulating resistin (Retn) in patients with HF is associated with miR-148b-3p expression, which suppresses the expression of Gadd45a and increases DNA damage, resulting in myocardial fibrosis, apoptosis and impaired cardiac function.37 Cao et al. narrowed down the genes associated with diastolic dysfunction in the HFpEF mouse heart transcriptome using a genome-wide association study of HF and identified Ascc2 as a DNA damage-reversal molecule. They showed that Ascc2 repression leads to Atm phosphorylation, increased Nppa expression and inflammatory responses, indicating that disruption of Ascc2-mediated DNA damage inhibition underlies the pathogenesis of HFpEF.38
Thus, this accumulating evidence reinforces the importance of DNA damage in HF and suggests the therapeutic potential of modifying this complex pathological process.
Heart Failure and Prognostic Factors
Brain natriuretic peptide (BNP) is probably the most clinically used marker for HF.39 Because HF is caused by a complex interplay of many pathological factors, including inflammation, oxidative stress and fibrosis, a variety of markers have been reported in addition to BNP. Interleukin 1 (IL-1), IL-6, tumour necrosis factor-α and highly sensitive C-reactive protein are known to be involved in inflammation, and for oxidative stress, oxidative low-density lipoprotein, myeloperoxidase and urinary biopyrrins have been reported. Cardiac troponin I and troponin T are useful markers of myocardial injury, which occurs in HF.40–42 In addition, cytokines and fibrosis markers, such as Igfbp7, resistin, osteoprotegerin and galectin 3, have also been noted as possible causes of HF.43–46
In our previous work, we also investigated whether we could predict the outcome of HF by assessing the degree of DNA damage in heart tissues using the DNA damage marker poly-ADP-ribose (PAR). Some patients recover from HF after adequate medical treatment, a phenomenon known as left ventricular reverse remodelling (LVRR). LVRR is correlated with a favourable prognosis for HF. We used myocardial biopsy specimens (collected at the time of diagnosis) from patients with HF due to dilated cardiomyopathy (DCM) and analysed the relationship between PAR positivity in cardiomyocytes and the incidence of LVRR. We found that patients with higher PAR positivity were less likely to develop LVRR and had poorer prognosis (Figure 2D). This indicates that DNA damage is also related to the reversibility and prognosis of HF in the clinical setting (Figure 2E).47
Genetics and Heart Failure
Advances in genetic engineering have enabled a better understanding of disease.48 In mice, knockout or overexpression of genes involved in cardiovascular diseases has enabled their function to be verified, thus advancing our understanding of the pathogenesis on a gene-by-gene basis. However, findings derived from experiments using mice are not always transferable to humans. Thus, by analysing the genomic DNA of patients and correlating genotypes with clinical and prognostic data, it is possible to deepen our understanding of the relationship between disease and genes to generate new stratifications of disease groups, and to predict outcomes. Expanding our knowledge by combining murine and human data is the key to medical genome science.49
Genetic Mutations and Heart Failure
Given that HF has various causes, there are only a limited number of cases in which a single molecular abnormality explains the pathophysiology. However, approximately 40% of DCM patients have a family history of DCM, suggesting the involvement of several gene factors.50 Although more than 100 genes have been reported to be causative factors for DCM, the relationship between mutation type and clinical course has not been fully elucidated.50 We performed panel sequencing of genetic variants in 120 Japanese patients with DCM and analysed the association between genetic variants and the clinical course.51 We found that titin-truncating variants (TTNtvs) were the most common (16.7%), and Lamin A/C (LMNA) mutations were the second most common (10.8%). Patients with TTNtvs responded to medical therapy and had a better prognosis, whereas those with LMNA mutations did not respond to existing treatments and had a poorer prognosis. The patient’s genetic information may be reflected in policy decisions for advanced therapies such as device implantation and heart transplantation. As the analysis of other HF causative diseases and the accumulation of genetic knowledge progresses, it is expected to lead to the realisation of precision medicine, in which genetic knowledge is applied to clinical decision-making.
Disease Stratification Based on Genetics
Gene sequencing technology has developed dramatically over the past two decades, enabling both genomic and transcriptomic information to be obtained inexpensively and rapidly, contributing to new biological insights.
Many previous studies have been limited to a few genes and intracellular signalling pathways that appear to be associated with disease phenotypes using gene expression data obtained from diseased animal organs or patient specimens. Recently, however, the reduction in the time and cost required to acquire transcriptome data has made it possible to follow the dynamics of comprehensive gene expression in organs at disease onset and during disease progression, even at the single-cell level. As a result, it is now possible to use longitudinal samples to infer the pathways of gene expression changes that lead to disease based on similarities in expression patterns.
Using a mouse model of HF, we investigated the gene expression patterns of cardiomyocytes by temporal single-cell analysis.52,53 Chronological analysis of murine hearts after pressure overload showed that pressure loading first caused compensatory hypertrophy of cardiomyocytes, followed by bidirectional bifurcation into compensated and failing states of cardiomyocytes, and myocardial remodelling during the development of HF. Analysis of the relationship between the degree of hypertrophy and gene expression in hypertrophied cardiomyocytes indicated activation of ERK1/2 and Nrf1/2 signals, which are involved in the hypertrophic response and antioxidant defence of the heart. We also analysed the gene network activated during the transition from hypertrophy to failure and found that p53 was an important regulator of cell fate conversion. This observation confirms that the cellular identity and morphological phenotypes of cardiomyocytes are encoded by a transcriptional program.53 Thus, by analysing gene expression at the turning points in cell fate, we can elucidate the molecular changes in tissues early in disease development, the heterogeneous processes of adaptation and disruption, and the factors that lead to a point of no return in disease progression.
A series of studies have been published on cardiac single-nuclei RNA sequencing (snRNA-seq) in patients with HF, which investigated the diversity of gene expression in patients, in disease aetiology, and in the heart of a single patient with HF. Koenig et al. performed snRNA-seq of DCM and healthy hearts and identified the major cardiac cell types and their specific transcriptional programs. They found cell type-specific transcriptional networks that regulate ageing and disease status. In the samples, the cardiomyocytes converged on a relatively common disease-related gene expression state, while fibroblasts and myeloid cells underwent dramatic disease-specific transcriptional diversification. In particular, the shift of fibroblasts to an activated state and the expression of CD14+ monocyte-derived inflammatory mediators were specific to failing hearts, suggesting that intervention in the cellular linkage network, including non-cardiomyocytes, may lead to novel therapeutic strategies.54
Chaffin et al. performed snRNA-seq of DCM, HCM and control human hearts, and described genetic alterations in failing hearts.55 In addition to constitutive changes in cell types between cardiomyopathy and non-failing hearts, they found common transcriptional pathways in cardiomyopathy patients, a decrease in proliferative CCR2-negative macrophages in the heart, and an increase in activated fibroblasts in the heart. Furthermore, in combination with CRISPR screening, they identified novel genes (PRELP and JAZF1) that contributed to fibroblast activation. This demonstrates the transcriptional diversity of healthy and diseased conditions in the human heart, as well as the shared molecular mechanisms involving non-cardiomyocytes and potential new therapeutic targets for HF.55
Reichart et al. used snRNA-seq in samples from patients with DCM and arrhythmogenic cardiomyopathy (ACM). They found that the cardiac cell atlas in DCM and ACM had different responses in the right and left ventricles, and that constitutive cardiac cell types, gene expressions, molecular signals and predicted intercellular communications differed according to the type of genetic mutation. This provides substantial evidence that genotype influences the pathological remodelling of cardiomyopathy and highlights heterogeneity in the cellular and molecular architecture of human HF. It reverses the common belief that HF converges on a common final pathway and provides guidelines for the future development of selectively targeted therapies to achieve personalised medicine.56
Conclusion
Many new drug and non-drug therapies have been developed for the treatment of HF in the past two decades. This process has been achieved through numerous clinical trials and post-marketing surveillance, and our field has been successful in further improving the prognosis of patients with HF. However, in order to address unmet needs, it is increasingly important to continue to update the existing clinical guidelines. One of the remaining challenges in HF management is the presence of non-responders to therapy. If the comprehensive genetic information-based practice described in this review becomes possible, diagnosis of the aetiology of HF may be more precisely subdivided, leading to the possibility of individualised and optimised treatment for each patient.
The other challenge is the achievement of preventive intervention at an earlier stage; prevention of HF requires interventions appropriate to each stage of disease progression. Such interventions include lifestyle modification to prevent the onset of the disease; treatment of lifestyle-related diseases such as hypertension and diabetes, which are established cardiovascular risk factors; appropriate treatment of underlying diseases such as ischaemic heart disease, arrhythmia and valvular disease; and maintenance therapy and rehabilitation to prevent transition to end-stage HF. As described above, we have proposed a new paradigm in the pathogenesis of HF, including the importance of myocardial ischaemia in the transition from cardiac hypertrophy to HF and the critical role of DNA damage and p53 activation in the progression of HF, focuses mainly on the treatment of the underlying disease and the prevention of transition to end-stage HF. Hence, the use of effective preventive measures at an earlier stage would be ideal from the standpoint of burden on medical resources. It is also desirable to broaden the scope of research in the fields of lifestyle modification and the treatment of lifestyle-related diseases in order to elucidate the mechanisms of HF even before its onset.
Basic research on HF is expected to improve the understanding of its pathogenesis and promote the development of new treatments for the next generation, thereby extending healthy life expectancies worldwide. 
Clinical Perspective
- The prognosis for heart failure (HF) has improved with the development of various therapies.
- To combat the pandemic of HF in the growing elderly population, it is necessary to establish novel methods of treatment and prevention based on an accurate understanding of pathogenesis. Therefore, basic research in this area is important.
- The accumulation of DNA damage and subsequent p53 activation play an important role in the transition from cardiac hypertrophy to functional failure, and these hallmarks are useful for predicting the therapeutic response.
- Understanding the disease at the molecular level is important for precise and accurate diagnosis. With the development of nucleic acid sequencing technology, it is now possible to examine gene mutations and single-cell gene expression in patients with HF. Disease reclassification and treatment decisions based on this information are expected to advance in the near future.